真核有参转录组测序
转录组广义上指特定细胞在某一功能状态下的所有转录产物��,包括mRNA 和非编码RNA(ncRNA)�����。是连接基因组遗传信息与生物功能的必然纽带��。真核生物转录组测序基于高通量测序��,可快速获得某一物种特定细胞或组织在某一状态下的所有转录本的集合���,用于研究基因结构和基因功能�����、可变剪接和新转录本预测等���。
转录组研究能够从整体水平研究基因功能以及基因结构���,已经成为揭示生物生长发育调控和逆境胁迫适应机制��、生物进化规律���、疾病发生发展的重要机制以及发现致病基因调控的关键靶点等方面的优先研究手段���,目前已广泛应用于基础研究���、临床诊断和药物研发���、动植物育种等各个领域���。
尊龙凯时组学优势
1.全流程服务���: 提供从生物样品到可视化数据的全套组学服务�����;
2.丰富项目经验���:具备处理复杂样本的能力��,同时具有丰富的建库经验���;
3.流程化分析��:完善的分析流程����,准确快速解析组学数据���;此外可结合客户的需求灵活进行定制化信息分析���;
4.专业化团队���:资深科学顾问具有丰富的方案设计����、售后服务经验���,全程一对一沟通��;
5.一站式解决方案���:拥有一站式多组学服务���,助力基础研究及临床治疗�����。
样本起始量与送样建议
|
样本类型 |
起始量 |
|
动物及临床脏器组织/脑组织等 |
>20mg |
|
动物及临床皮肤/骨/血管/脂肪组织等 |
>100mg |
|
植物叶片组织/花 |
>200mg |
|
植物根/茎/果实/种子 |
>500mg |
|
原代细胞/细胞系 |
>5×106个 |
|
中性粒细胞/嗜酸性粒细胞/嗜碱性粒细胞 |
>5×107个 |
|
总RNA |
>1μg且RIN>7.0 |
注意事项���:
① 组织样本建议保存在RNAlater�����、RNAHold��、RNAProtect等相关组织保存液中����,然后-80℃保存或干冰寄送����;
② 细胞样本使用TRIzol等裂解液充分裂解之后��,-80℃保存或干冰寄送��;
③ 更加详细的样本准备指南����,请联系在线客服���。
生物信息学分析内容
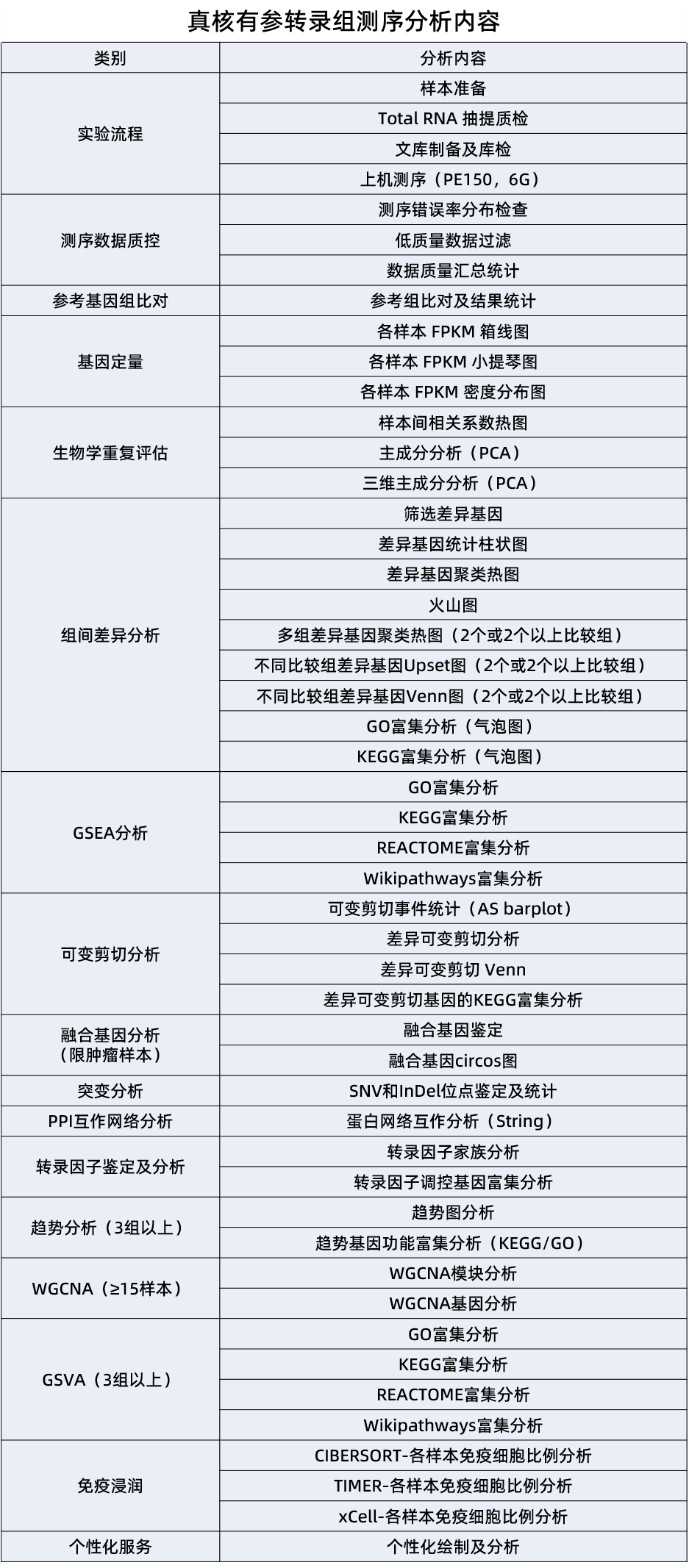 应用场景
应用场景
应用场景1����:差异基因筛选与功能分析
适用范围�����:临床医学��、基础医学���、生物化学����、动植物及真菌研究等任意方向
利用真核有参转录组测序�����,可通过比较实验组和对照组基因表达量筛选差异表达的基因��。然后对差异表达基因进行进一步锁定���,如通过GO���、KEGG富集分析及GSEA分析���,配合Pubmed中已发表的文献以及课题组中已积累的部分明星分子对差异表达基因进行功能注释���,并进一步分析关注的功能基因��。进入实验验证阶段后可对筛选到的差异基因进行qPCR��、Northern���、Western Blot���、FISH验证���、基因敲除及过表达等����。
应用场景2���:时序分析或浓度梯度分析
适用范围�����:临床样本���、细胞样本���、动植物样本有多个时间段的样本��,或不同药物浓度处理的样本
在转录组数据分析过程中���,有一类特殊的实验设计����。通过对不同时间段的实验样本进行搜集����,或测试不同的药物���、试剂等浓度梯度的样本进行采集����。继而研究不同基因在不同时间段或不同浓度梯度间的表达规律���,这一类分析通常称之为“时序分析”
应用场景3����:转录因子/调控因子/剪切因子等上游调控基因挖掘
适用范围��:临床医学���、基础医学����、生物化学����、动植物研究等任意研究方向
常规转录组差异分析极有可能得到大量的差异基因��,这对后期实验验证的目标锁定带来挑战��。在没有特定感兴趣的通路及明星分子前提下��,转录因子是一个非常不错的切入方向�����。转录因子可以调节基因组DNA开放性����、募集RNA聚合酶进行转录过程���、募集辅助因子调节特定的转录阶段����,调控诸多生命进程���,诸如免疫反应��、发育模式等����。所以��,分析转录因子表达及其调控活性对于解析复杂生命活动具有重要意义����。其他调节因子包括可变剪切等调控基因也可以参与上游调控����。
应用场景4���:大样本研究
适用范围���:动植物育种���、遗传群体与物种起源���、人群队列与生物标志物挖掘
随着测序技术的飞速发展����,少量样本的转录组测序研究已经无法解释复杂的生物学问题����。研究者们已开始利用大样本量的转录组样本���,结合统计学与机器学习等方式���,知道符合特定规律和研究目的的核心基因���。如孟德尔随机化��、相关性分析�����、线性回归�����、LASSO回归����、Cox回归等���,分析不同样本基因或基因组多样性��,挖掘更深入和全面的生物学意义���。
项目文章
【1】Liang H, et al. Siglec15 facilitates the progression of non-small cell lung cancer and is correlated with spinal metastasis. Ann Transl Med. 2022 Mar;10(6):281. PMID: 35434017.
【2】Pi Y, et al. The role of PIWI-interacting RNA in naringin pro-angiogenesis by targeting HUVECs. Chem Biol Interact. 2023 Feb 1;371:110344. PMID: 36623717.
LncRNA测序
长链非编码RNA(lncRNA)是一类长度大于200 nt 的非编码RNA(ncRNA)���,广泛存在于各种生物体内���,在表观遗传调控����、细胞周期调控和细胞分化调控等众多生命活动中发挥重要作用�����,与动植物的生长发育����,人类的疾病发生有着密切关系����,也可作为疾病诊断的标志物或是重要靶点���。
利用高通量测序技术进行lncRNA 测序及生物信息学分析����,可快速准确地发现那些具有重要调控功能的lncRNA��,分析其与特定生物学过程的关系�����,深入探索lncRNA的功能及其表达调控机制���。包括竞争结合miRNA的ceRNA调控机制��,及对染色质结构进行一系列调控如增强子����、启动子��、绝缘子等���。
技术优势
1. 文库优化���:采用核糖体去除和链特异性文库构建方案��,保留了完整的lncRNA和mRNA序列�����,以及序列方向性���。
2. 序列完整�����:对样本中几乎全部的lncRNA和mRNA序列进行鉴定和分析����。
3. 物种多元���:对包括人类��、动植物在内的许多物种的lncRNA进行鉴定和分析�����。
4. 分析严谨����:系统收集lncRNA数据库用于鉴定已知lncRNA��,并设置严格的筛选条件用于发现新lncRNA��。
5. 关联全面���:开展lncRNA与mRNA的关联分析���,深入分析lncRNA调控网络��。
样本起始量与送样建议
|
样本类型 |
起始量 |
|
动物及临床脏器组织/脑组织等 |
>20mg |
|
动物及临床皮肤/骨/血管/脂肪组织等 |
>100mg |
|
植物叶片组织/花 |
>200mg |
|
植物根/茎/果实/种子 |
>500mg |
|
原代细胞/细胞系 |
>5×106个 |
|
中性粒细胞/嗜酸性粒细胞/嗜碱性粒细胞 |
>5×107个 |
|
外泌体样本 |
>1×108个 |
|
血清/血浆/脑脊液/关节积液/卵泡液 |
>2mL |
|
细胞培养上清液 |
>20mL |
|
尿液 |
>30mL |
|
总RNA |
>1μg且RIN>7.0 |
注意事项���:
① 组织样本建议保存在RNAlater��、RNAHold�����、RNAProtect等相关组织保存液中�����,然后-80℃保存或干冰寄送���;
② 细胞样本使用TRIzol等裂解液充分裂解之后���,-80℃保存或干冰寄送
③ 更加详细的样本准备指南��,请联系在线客服
应用场景
应用场景1����:启动子/增强子调控
适用范围����:非线性转录调控���、新顺式作用元件发现及功能鉴定��、组织特异性启动子筛选及确定等研究
lncRNA的自身转录会干扰其邻近编码蛋白的基因的转录�����。上游lncRNA转录时��,会穿越邻近靶基因的启动子区���,干扰了转录因子与靶基因的启动子结合����,从而抑制靶基因的转录����。此外�����,lncRNA具有促进增强子环化和激活基因表达的功能����。在没有成环的情况下增强子处于非活性状态�����。
应用场景2���:与蛋白修饰/DNA甲基化/m6A联合
适用范围����:临床医学��、基础医学��、植物遗传育种等研究
lncRNA参与表观调控���,可能是招募一些染色质重构复合体来介导基因沉默���,尤其是一些与组蛋白修饰相关的组蛋白甲基转移酶的调控����。此外����,lncRNA还能与一些DNA甲基化和去甲基化酶结合使得基因的启动子区发生甲基化的变化从而影响基因表达��。
应用场景3����:ceRNA调控机制
适用范围��:环境应激���、临床医学���、植物遗传育种����、疾病早期诊断等研究
ceRNA全称competing endogenous RNA����,是一种能够竞争结合RNA的作用元件����。通常lncRNA和circRNA会竞争结合miRNA����,我们一般把lncRNA和circRNA可以称作ceRNA��。ceRNA调控网络全称ceRNAregulation network��,指的是有ceRNA参与的整个调控网络cascade����。而ceRNA分析指的是对整个ceRNA调控网络进行分析���。一般有circRNA-miRNA-mRNA分析或lncRNA-miRNA-mRNA分析���。
结果展示
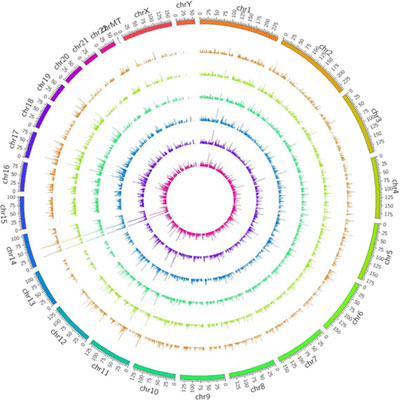
不同样本lncRNA基因组可视化结果为了更直观的展示lncRNA candidate在染色体中分布情况�����,我们采用circos(ww.circos.ca)软件对筛选获得的lncRNA进行基因组mapping���。
主要分为两部分进行展示���,一部分按照lncRNA的不同分类进行基因组mapping����,另一部分按照不同样本中lncRNA进行基因组mapping����。作图时分别将每条染色体按照每25mb为基本单位���,不同样本中lncRNA基因组可视化作图时统计每个区段中lncRNA的表达量绘图���,不同lncRNA类型基因组可视化时统计每个区段内lncRNA的数目绘图����。
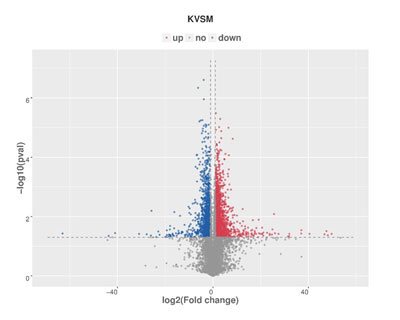
1.差异基因火山图
通过绘制火山图可以了解差异表达基因的整体分布情况���。以log2 (foldchange)为横坐标����,-log10(pvalue)为纵坐标���,对差异表达分析中所有的基因绘制火山图����。 其中横坐标代表基因在不同样本中差异表达倍数变化���;纵坐标代表基因表达量变化差异的统计学显著性���;up颜色代表上调的显著差异表达基因���,down颜色代表下调的显著差异表达基因��,no代表非显著性差异表达基因��。
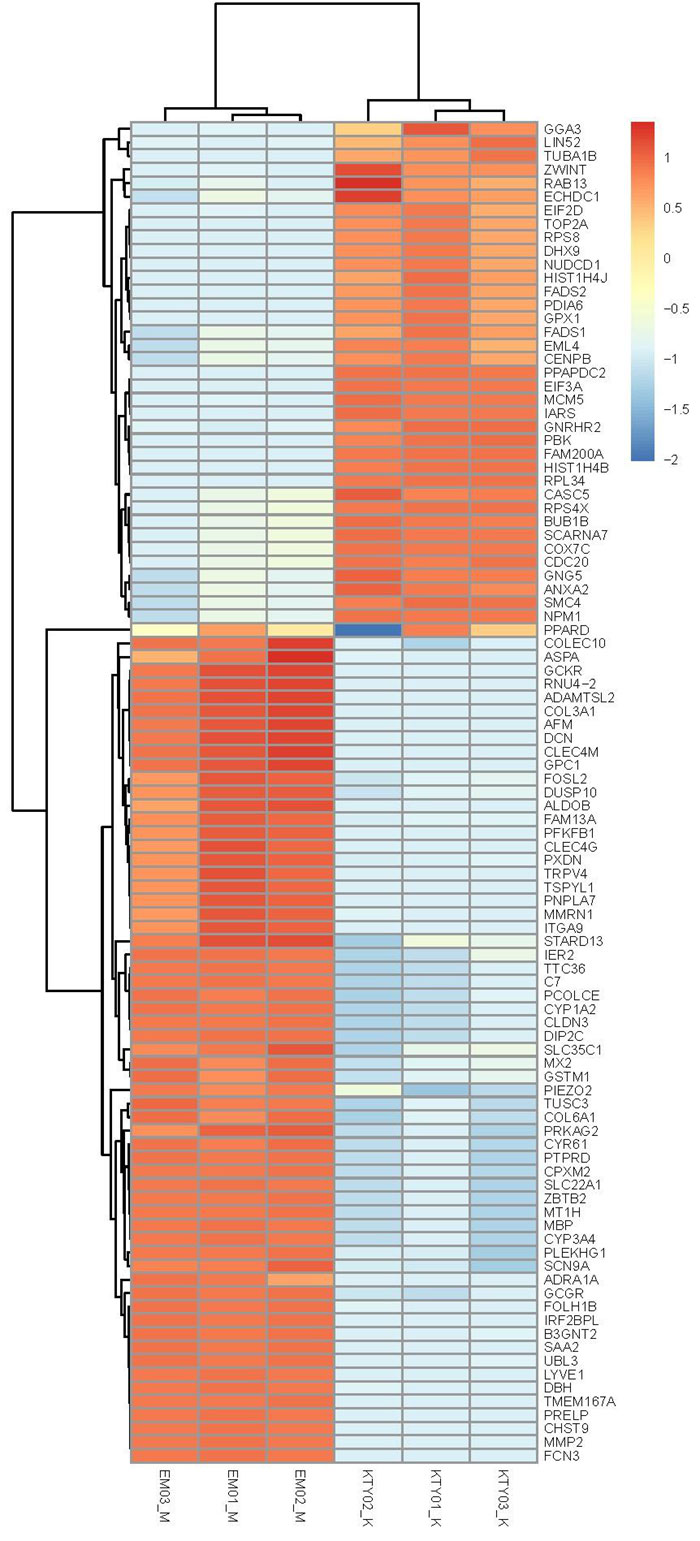
2.差异基因聚类热图
差异基因聚类分析用于判断基因在不同实验条件下调控模式的聚类模式���。根据样品基因表达谱的相近程度��,将基因进行聚类分析����,直观地展示基因在不同样品(或是不同处理)中的表达情况���,由此获取生物学相关信息����。为了更好的直观反映聚类表达模式���,对于非生物学重复
我们采用log10(FPKM+1)进行展示���,对于生物学重复将差异基因FPKM通过Z值方式进行基因表达展示����。其中横坐标为样本��,纵坐标为基因��,不同的颜色表示不同的基因表达水平���。
常见问题
1.什么是长链非编码RNA���?它们的作用有哪些��?
哺乳动物基因组序列的约5%~10%被稳定转录����,蛋白质编码基因仅约占1%���,其余4%~9%的序列都转录为非编码RNA��。而非编码RNA(non-coding RNA) 是指不能翻译为蛋白的功能性RNA分子��。长链非编码RNA为这4%~9%中长度大于200nt的非编码RNA��。它们的作用主要体现在四个方面�����:第一���,影响周边基因的表达��;第二���,调控蛋白质活动及定位���;第三�����,产生小分子RNA���;第四����,对其他RNA的调控作用��。
2.lncRNA测序为什么需要构建链特异性文库����?
很多基因组区域具有正负链的转录本�����,反义转录是真核基因的一个特征��,是一种重要的调控方式���。使用ssRNA-Seq(Strand-Specific)能够保留RNA的方向性信息���,可以确定转录本来自正链还是负链���,以便更加准确的获得基因的结构以及基因表达信息����。
3.预测lncRNA与mRNA����、miRNA互作的方法����?
1)cis调控预测����,筛选的条件只有cis locations是100kb���。(同一条染色体上距离lncRNA100kb的mRNA���,都会被列入到cis调控的可能)
2)trans调控预测用的是RIsearch软件��,是以最小自由能小于-11来筛选碱基互补配对的lncRNA与mRNA���。
3)在预测lncRNA与miRNA互作的时候���, 用score罚分值来判定��,一般≤4就认定为可信���,数值越小越可信����。
CircRNA测序
共价闭合�����、单链环状的RNA(circRNA)是一类比较特殊的RNA����,其没有游离的5′帽子结构和3′ poly(A)结构���,并且对核酸酶不敏感���,因此比普通的线性RNA(linear RNA)更稳定��。随着高通量测序以及生物信息分析技术的快速发展��,在不同物种中鉴定出成千上万个circRNA���,其中大多数circRNA 在不同物种间具有保守性和稳定性����,并且circRNA 的表达具有细胞特异性��、组织特异性以及发育阶段特异性���。circRNA除了常规的对miRNA竞争结合的ceRNA调控机制外��,还有翻译多肽等新功能����,在医学及动植物基础研究及后期转化上有着巨大的潜力��。
样本起始量与送样建议
|
样本类型 |
起始量 |
|
动物及临床脏器组织/脑组织等 |
>20mg |
|
动物及临床皮肤/骨/血管/脂肪组织等 |
>100mg |
|
植物叶片组织/花 |
>200mg |
|
植物根/茎/果实/种子 |
>500mg |
|
原代细胞/细胞系 |
>5×106个 |
|
中性粒细胞/嗜酸性粒细胞/嗜碱性粒细胞 |
>5×107个 |
|
外泌体样本 |
>1×108个 |
|
血清/血浆/脑脊液/关节积液/卵泡液 |
>2mL |
|
细胞培养上清液 |
>20mL |
|
尿液 |
>30mL |
|
总RNA |
>1μg且RIN>7.0 |
注意事项����:
① 组织样本建议保存在RNAlater����、RNAHold���、RNAProtect等相关组织保存液中����,然后-80℃保存或干冰寄送����;
② 细胞样本使用TRIzol等裂解液充分裂解之后���,-80℃保存或干冰寄送���;
③ 更加详细的样本准备指南���,请联系在线客服
应用场景
应用场景1���:结构分子招募核心蛋白与核酸
适用范围���:基础医学����、生物化学与分子生物学等任意研究方向
lncRNA和circRNA有时候会作为一种特殊的RNA分子��,形成蛋白-蛋白复合物或蛋白-核酸的骨架分子���,对下游分子行使一系列调控作用���。如部分circRNA会招募一些泛素化酶对下游的蛋白行使降解功能�����,或者部分lncRNA会招募一些转录因子与目的基因的启动子区域结合激活下游的基因表达等����。
应用场景2�����:外泌体
适用范围��:临床与转化医学���、基础医学等任意研究方向
lncRNA在外泌体中由于以碎片形式存在���,这种随机片段不仅不利于检测���,而且较低的浓度和较差的重复性����,使得lncRNA并不是外泌体研究的热门分子之一����。但是circRNA由于其稳定性�����,除了从体液样本外泌体中分离circRNA可作为疾病标志物外��,还能用于后期的外泌体治疗等领域�����,是后ceRNA时代中较为热门的circRNA细分研究领域方向之一��。其中一个方向是疾病标志物����,一般以大样本队列研究居多��。一种是后期转化及机制研究方向���,包括外泌体受体细胞分子机制研究以及疾病治疗等方向����。
应用场景3���:翻译多肽
适用范围��:临床与转化医学���、基础医学���、生物化学与分子生物学等任意研究方向
circRNA上有一段未翻译的RNA序列����,称为IRES����,它可以折叠成类似于起始tRNA的结构��,并招募更多的核糖体���。正常情况下����,IRES不与eIF结合�����,而是与一类称为ITAF的蛋白质结合��。ITAF的作用是将核糖体募集到circRNA的内部结构���,以启动蛋白质翻译���。此外���,还可能存在IRES增强子��,类似于circ-ZNF609的UTR元件����。也有研究通过翻译组测序与转录组测序��,发现了非编码RNA LINC-PINT形成环状RNA后可被翻译形成功能多肽��。
应用场景4�����:ceRNA调控机制
ceRNA全称competing endogenous RNA��,是一种能够竞争结合RNA的作用元件��。通常lncRNA和circRNA会竞争结合miRNA����,我们一般把lncRNA和circRNA可以称作ceRNA��。ceRNA调控网络全称ceRNAregulation network����,指的是有ceRNA参与的整个调控网络cascade�����。而ceRNA分析指的是对整个ceRNA调控网络进行分析����。一般有circRNA-miRNA-mRNA分析或lncRNA-miRNA-mRNA分析�����。
案例展示
circRNA和母基因通过不同的pathway影响肝癌细胞的生长����,迁移和侵袭
1.研究背景
肝细胞癌(HCC)是最常见的一种恶性肿瘤��,同时�����,中国也是肝癌的癌症大国���。虽然现在的医疗诊断技术发展很快����,但是����,对与肝癌的现有诊断技术还是受限很大���。因此���,开发新的诊断biomarker�����,并理解这些biomarker的作用机制����,是很多科研人员想做的事情��。本研究从circRNA���,一类非常适合做biomarker的候选分子出发����,深入研究了一个锌指家族基因ZKSCAN1及其产生的circZKSCAN1���,在HCC中的作用机制及作为biomarker的潜在价值����。
2.研究结果
1)基因表达模式分析
ZKSCAN1�����,是一个锌指蛋白家族���,该家族基因在肿瘤发生��,发展����,转移和药物耐受方面都发挥着非常重要的作用����。同时��,现有的研究也发现���,ZKSCAN1基因的第二个和第三个外显子部分能形成一个circRNA��,在人脑部和肝脏表达丰富���。因此��,研究人员首先验证了ZKSCAN1和circ ZKSCAN1在HCC组织中的表达情况���。102个HCC组织和配对癌旁的RT-PCR结果显示��,他们在HCC样本中的表达显著降低��。进一步将ZKSCAN1和circ ZKSCAN1的表达与临床指标进行关联分析��,发现低的ZKSCAN1表达与肿瘤大小显著相关��;而circ ZKSCAN1的表达与不同肿瘤数目���,硬化程度����,血管侵袭��,微血管侵袭���,肿瘤grade都显著相关��。ROC曲线分析显示��,circ ZKSCAN1的AUC值达到0.834����,敏感性和特异性能达到82.2%和72.4%��,相比而言���,ZKSCAN1的AUC值只有0.474���。因此��,circ ZKSCAN1的表达更合适作为肿瘤组织的诊断biomarker��。
2)circRNA与母基因表达相关性分析
对于circRNA的研究�����,第一个想到的是circRNA的表达是否会和母基因表达有关联���。因此����,研究人员先在不同人HCC细胞系中检测了ZKSCAN1 mRNA和circZKSCAN1的表达����。发现所有的细胞系(Huh7, SMMC-7721, BEL-7402, HepG2和HepG3B)相比对照组(L02)��,表达都降低��。通过在HepG2和SMMC7721中过表达和敲降ZKSCAN1和circZKSCAN1��,研究人员发现过表达或者敲降其中一个����,不会影响另一个的表达变化���。因此����,ZKSCAN1 mRNA和circZKSCAN1的表达彼此独立��,不相互影响���。
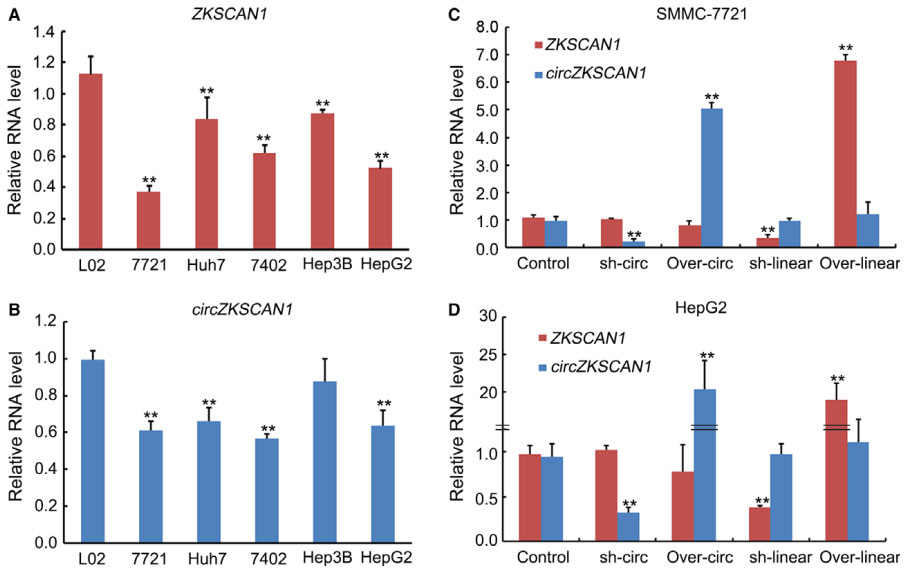
3)基因功能分析
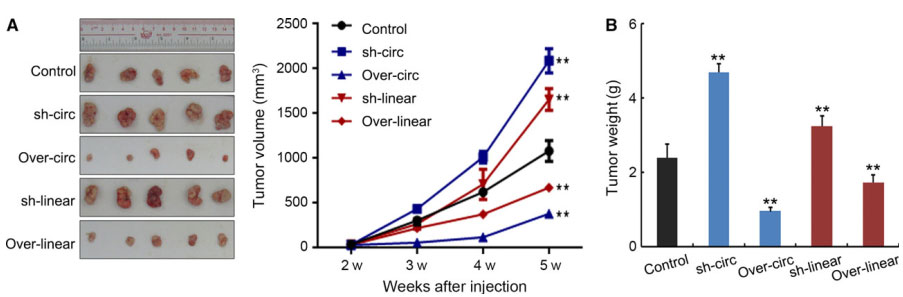
为探究ZKSCAN1 mRNA和circZKSCAN1对HCC细胞的功能影响����,研究人员进行了相关检测��。结果显示���,在ZKSCAN1 mRNA和circZKSCAN1的敲降细胞中��,增殖能力显著增强��,而过表达细胞中���,显著下降����;过表达细胞的迁移和侵袭能力显著降低���;敲降细胞显著增加���。进一步的裸鼠种植实验也表明����,过表达时肿瘤生长抑制���,而敲降时刺激肿瘤生长����。因此���,ZKSCAN1 mRNA和circZKSCAN1对肿瘤生长有显著影响����。
4)基因亚细胞定位
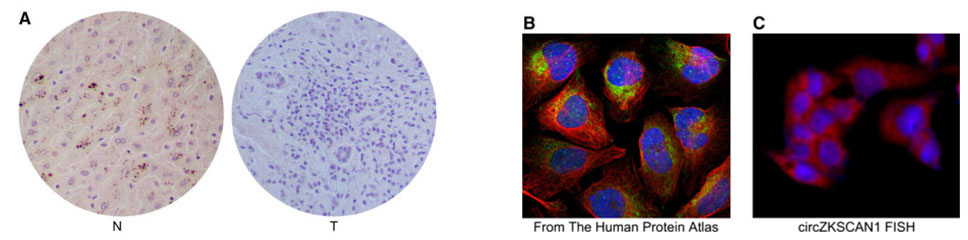
基因在细胞中的定位往往决定着他们的功能行使����。因此��,研究人员对ZKSCAN1的蛋白进行亚细胞定位����。结果显示����,正常肝组织的细胞质中存在大量的ZKSCAN1����,而HCC肝组织中却很少��。另外���,FISH分析表明circ ZKSCAN1也是主要定位于细胞质中����。
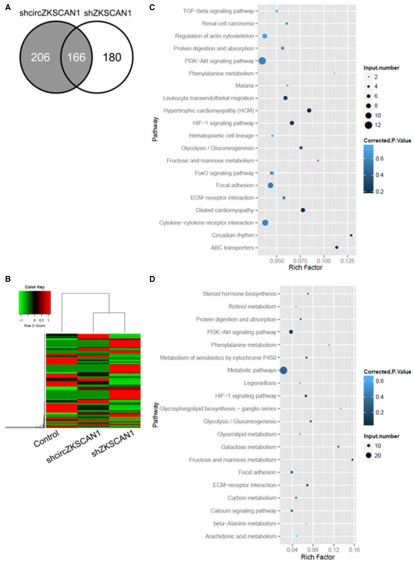
5)下游调控基因分析
为探究ZKSCAN1调控细胞增殖和侵袭的内在分子机制����。研究人员对敲降ZKSCAN1 mRNA和circ ZKSCAN1的HCC细胞与对照进行了全基因组RNA测序�����。结果显示���,敲降circZKSCAN1引起372个基因差异表达��;敲降ZKSCAN1引起346个基因差异表达����。其中��,两组结果中共有166个是在两种情况下都出现差异��。KEGG通路分析显示����,敲降circ ZKSCAN1的差异基因富集到PI3K pathway, migration pathway, actin cytoskeleton pathway等����;而敲降ZKSCAN1则主要富集到metabolic pathways���。因此�����,circ ZKSCAN1和ZKSCAN1影响了下游不同的信号通路影响细胞增殖和侵袭����。
6)qRT-PCR验证下游基因
最后���,研究人员对特异性地在ZKSCAN1敲降后变化的基因与circZKSCAN1敲降后变化的基因及都变化的基因进行了qRT-PCR验证���,结果表明����,在ZKSCAN1敲降后���,细胞代谢相关基因确实发生了显著变化���;而在circZKSCAN1敲降后����,凋亡和迁移相关基因发生显著变化���;COL3A1��,CDH5��,MYB����,PDK1和BCL2在两种情况下都显著变化����。
研究结论
在本研究中�����,研究人员发现�����,虽然ZKSCAN1和circRNA在HCC中都发生了显著下调��,都是影响细胞生长��,迁移和侵袭���,但是�����,他们影响的下游信号通路却是各不相同��。该研究为理解circRNA和其母基因在生命体中的调控作用提供了新的依据����。
参考文献
Yao Z, Luo J, Hu K�����,et al.(2017)ZKSCAN1 gene and its related circular RNA (circZKSCAN1) both inhibit hepatocellular carcinomacell growth, migration and invasion but through different signaling pathways. Mol Oncol. doi: 10.1002/1878-0261.12045
结果展示
1.基因表达值密度
不同样本各自表达量处理后数值log10(fpkm)做表达值密度图�����,可以比较不同样本间表达趋势的变化���。理想状态下��,各样本的表达密度图应符合正态分布��,同时生物学重复样本的表达趋势应趋于一致����。
2.差异circRNA-hosting gene GO富集性柱状图
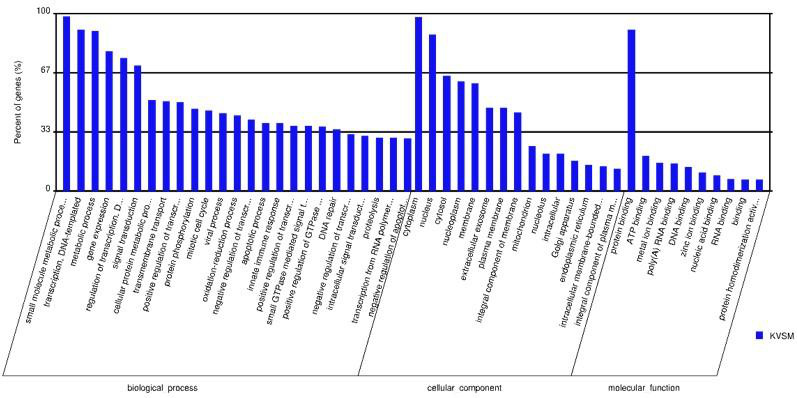
GO富集性分析结果柱状图反映在生物过程(biological process)����、细胞组分 (cellularcomponent)和分子功能(molecular function)富集的GO term上差异基因的个数分布情况���。
3.差异circRNA-hosting gene KEGG富集性散点图
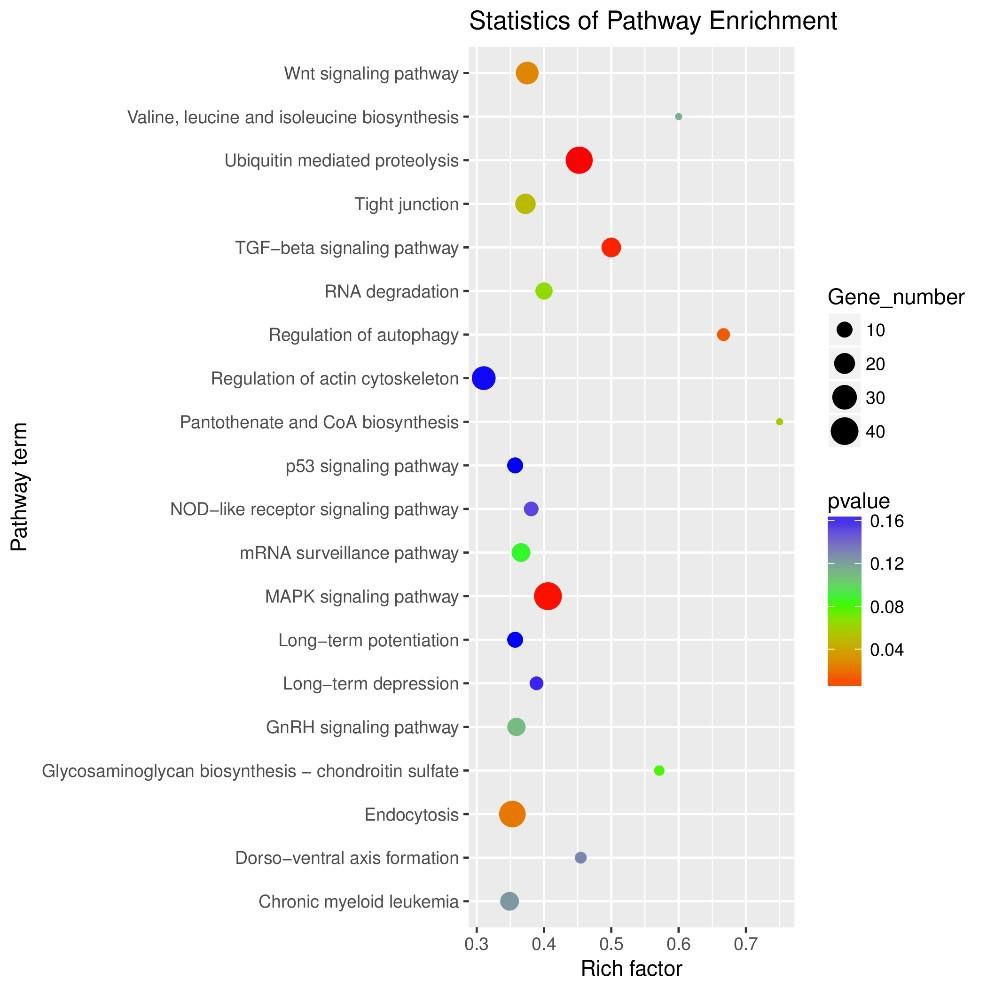
通过ggplot2对KEGG富集分析结果以散点图的形化进行展示��,其中横坐标Rich factor表示位于该KEGG的差异基因个数/位于该KEGG的总基因数(Rich factor=S gene number/B gene number)��,Rich factor越大����,KEGG富集程度 越高�����。纵坐标是Pathway term����,即KEGG代谢通路����。散点图中����,点的大小代表 S gene number 匹配到单个KEGG的具有显著性差异的基因数���,点的颜色代表 富集分析的p值���,即富集的显著性���。KEGG富集分析散点图是根据富集的显著 性(pvalue)取top20的Pathway term进行绘图���。
常见问题
1.CircRNA的成环机制都有哪些��?
CircRNA的成环机制有��:套索驱动的环化��;内含子配对驱动的环化��;内含子环化����;RBP或一类反式因子驱动的环化���;可变剪接引起的环化����。
2.circRNA测序和普通的RNA-seq���,在抽提total RNA时有区别吗����?
circRNA测序对total RNA的要求与普通的RNA-seq相同��,所以抽提方法也相同����。但是由于circRNA建库需要去除rRNA和线性RNA��,所以需求大量的total RNA����,需提供大于等于10 μg的total RNA����。
3.circRNA与lncRNA的区别是什么��?
结构上��,circRNA没有自由的5’端和3’端����;功能上����, circRNA功能研究比较单一���,现在对于其是否能够像lncRNA一样在染色体水平����、蛋白质水平上具有调控作用��,尚未可知��,这些也是现在研究的方向����。根据已有研究发现�����, circRNA可以发挥海绵效应吸附miRNA��,可以作为竞争性内源RNA��。
Small RNA测序
Small RNA主要包括miRNA���、piRNA��、tsRNA(tRF&tiRNA)���、snRNA和snoRNA等����,其中miRNA研究最为广泛���,是一类不具有蛋白编码能力的RNA分子��,能调控基因表达����,在细胞生长���、发育和代谢等基础生物学过程中都扮演着重要的角色���,甚至在癌症等相关疾病形成过程中也起着关键的作用����。
尊龙凯时组学服务优势
1. 高度灵活性��:可研究任一 17-35 nt 之间的小 RNA 分子
2. 高度准确性���:可准确到单核苷酸水平����,有利于区分高度同源的小 RNA 分子和鉴定小 RNA 的多态性
3. 检测动力学范围广��:可在超过 6 个数量级范围内实现准确的序列检测和定量分析���,有利于低丰度小 RNA 差异表达的研究
4. 数据质量高���:深度测序保证了抽样随机性��、可靠性和重复性���,与实时定量 PCR 结果具有较高的一致性
样本起始量与送样建议
|
样本类型 |
起始量 |
|
动物及临床脏器组织/脑组织等 |
>20mg |
|
动物及临床皮肤/骨/血管/脂肪组织等 |
>100mg |
|
植物叶片组织/花 |
>200mg |
|
植物根/茎/果实/种子 |
>500mg |
|
原代细胞/细胞系 |
>5×106个 |
|
中性粒细胞/嗜酸性粒细胞/嗜碱性粒细胞 |
>5×107个 |
|
外泌体样本 |
>1×108个 |
|
血清/血浆/脑脊液/关节积液/卵泡液 |
>2mL |
|
细胞培养上清液 |
>20mL |
|
尿液 |
>30mL |
|
总RNA |
>1μg且RIN>7.0 |
注意事项���:
① 组织样本建议保存在RNAlater���、RNAHold����、RNAProtect等相关组织保存液中�����,然后-80℃保存或干冰寄送��;
② 细胞样本使用TRIzol等裂解液充分裂解之后���,-80℃保存或干冰寄送
③ 更加详细的样本准备指南���,请联系在线客服
应用场景
应用场景1���:外泌体
适用范围��:临床与转化医学�����、基础医学�����、动物/兽医研究等任意研究方向
MicroRNA(miRNA)是一类非编码RNA��,能够调节一系列广泛的生物过程���。除了在细胞内发挥作用外����,最近研究还显示miRNA在细胞外泌体中分泌�����,从而允许其转移至近端或远端的细胞进而调节基因表达����。外泌体代表着有希望可以从中分离miRNA的纳米材料���,有发挥治疗作用的潜力����。
应用场景2�����:大样本研究
适用范围���:临床与转化医学��,疾病标志物挖掘与鉴定
miRNA是一种稳定性强重复性好易于检测的非编码RNA���,在体液样本及组织样本中丰度高��,在临床上具备成为特异性强灵敏度的生物标志物(biomarker)的潜能�����。例如外泌体或体液样本中����,超过70%的RNA为miRNA���,配合回归和各类机器学习建模��,可挖掘高灵敏度的潜在生物标志物�����。
应用场景3����:抑制翻译/激活翻译
适用范围���:临床与转化医学����、基础医学与分子生物学����、动植物研究等任意研究方向
miRNA可通过抑制核糖体的组装来阻断翻译起始����,进而起到对翻译过程的抑制作用��。miRNA的抑制作用需要靶mRNA具有m7G帽子结构成为支持这一理论的重要依据���,由此可以推断 miRISC可能通过对翻译起始复合物形成抑制而发挥作用���;Ago2中间结构域具有结合m7G帽子的活性���,Ago2通过 对miRNA招募靶mRNA的 3' UTR��,从而与起始复合物eIF4E/G竞争性结合m7G帽子���,最终发挥对翻译起始复合物的抑制作用����。实际上miRNA具有双重功能��,当位于胞浆时可抑制基因表达���,当位于细胞核内可激活基因的转录��。核内miRNA可通过结合增强子��,改变增强子的染色质状态��,从而激活基因的转录表达���。
应用场景4���:与单细胞测序联合分析
适用范围���:临床与转化医学��、基础医学与分子生物学���、动植物研究等任意研究方向
miRNA与单细胞联合分析���,主要通过两种方法���。第一种是通过对miRNA的靶基因所对应的基因集在单细胞测序中用AddModuleScore打分模块实现���。第二种就是引入了RIP和CLIP数据库来进行后期校正���。即间接打分和直接打分两种方法���。
通过miRNA与单细胞联合��,能够锁定通路富集较高的细胞亚群����,并和miRNA建立起直接调控和间接调控两种方式��。
应用场景5����:ceRNA调控机制
适用范围���:临床与转化医学���、基础医学与分子生物学��、动植物研究等任意研究方向
ceRNA全称competing endogenous RNA��,是一种能够竞争结合RNA的作用元件���。通常lncRNA和circRNA会竞争结合miRNA���,我们一般把lncRNA和circRNA可以称作ceRNA���。ceRNA调控网络全称ceRNAregulation network����,指的是有ceRNA参与的整个调控网络cascade���。而ceRNA分析指的是对整个ceRNA调控网络进行分析����。一般有circRNA-miRNA-mRNA分析或lncRNA-miRNA-mRNA分析���。
应用场景6�����:植物miRNA与降解组联合
适用范围��:植物抗病抗旱等抗逆研究����、植物遗传育种���、植物发育学等任意研究方向
通过降解组测序和miRNA测序的联合应用����,研究者不仅可以系统鉴定多种植物中miRNA调控的靶基因���,还可以探究多种miRNA及其靶基因对植物不同处理的差异性表达��,证实某些miRNA的靶基因在植物生长发育过程中的重要作用���。
项目文章
【1】Su T, et al. Bone Marrow Mesenchymal Stem Cells-Derived Exosomal MiR-29b-3p Regulates Aging-Associated Insulin Resistance. ACS Nano. 2019 Feb 26;13(2):2450-2462. PMID: 30715852.
【2】Ma L, et al. Nanotopography Sequentially Mediates Human Mesenchymal Stem Cell-Derived Small Extracellular Vesicles for Enhancing Osteogenesis. ACS Nano. 2022 Jan 25;16(1):415-430. PMID: 34935354.
结果展示
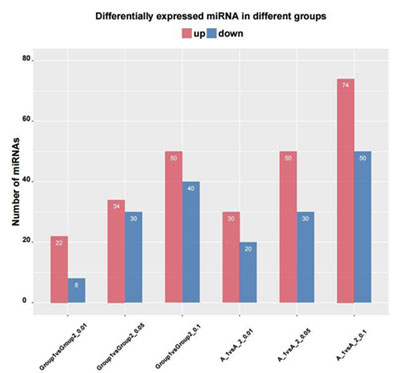
1.差异miRNA上下调统计分析
差异基因上下调频数统计用于判断不同实验条件下差异表达miRNA的个数���。其中横坐标表示比较组信息��,纵坐标表示上下调miRNA的数目����,红色代表上调miRNA���,蓝色代表下调miRNA����,其中数字代表上下调miRNA的数目�����。
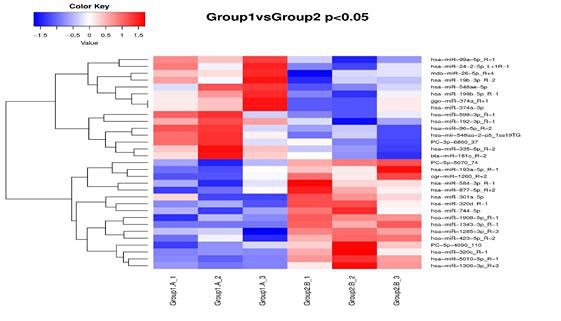
2.差异miRNA聚类分析
差异miRNA聚类分析用于判断miRNA在不同实验条件下调控的聚类模式���。根据样品miRNA表达谱的相近程度����,将miRNA进行聚类分析����,直观地展示miRNA在不同样品(或是不同处理)中的表达情况����,由此获取生物学相关信息���。不同的颜色表示不同的miRNA表达水平���,颜色由蓝色经由白色至红色表示表达量从低到高���。红色表示高表达基因��,深蓝色表示低表达基因���。
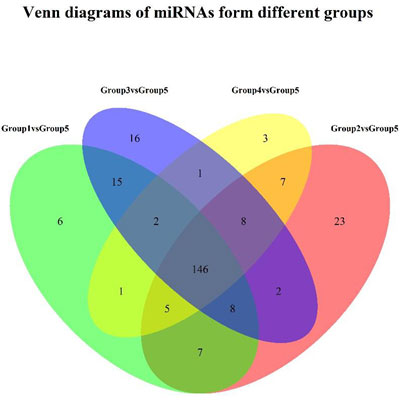
3.差异miRNA韦恩图
通过不同比较组之间差异基因韦恩图可以直观地显示出不同比较组之间共同的和特有的差异表达miRNA的个数��,差异基因韦恩图具有明显的生物学意义���,比如是相同对照不同处理的实验设计情况���,可以将不同处理下的差异基因进行比较��。
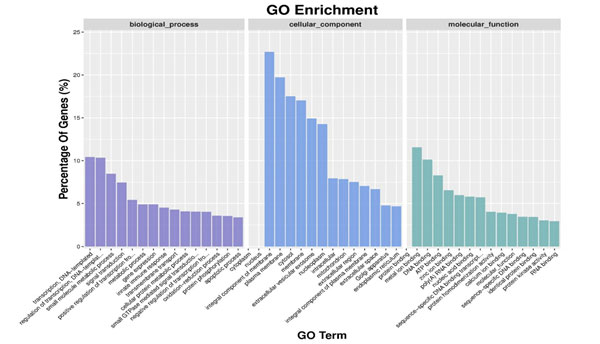
4.GO富集性柱状图
miRNA靶基因的GO富集柱状图���:用于反映在生物过程(biological process)��、细胞组分(cellular component)和分子功能(molecular function)富集的GO term上差异基因的个数分布情况����。
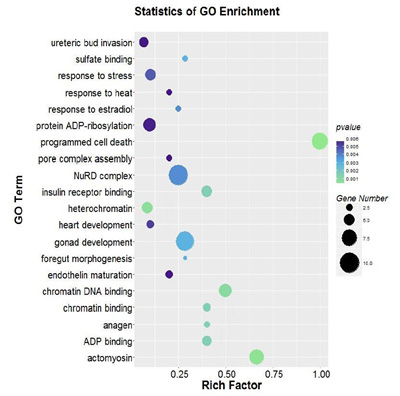
5.GO富集性散点图
对差异miRNA进行GO富集分析并以散点图展示����,Rich factor表示位于该GO的差异基因个数/位于该GO的总基因数���,P值越小���,GO富集程度越高�����。
常见问题
1.miRNA命名规则是怎样的���?
关于miRNA命名���,是根据miRBase的命名规则���:物种拉丁名3字母缩写-miR/MIR-编号(植物)���,miR表示的是microRNA成熟体��,植物的前体使用MIR����。参见miRBase官网:目前已经不再使用*来标记microRNA与其发夹前体互补配对位置的互补序列��,而是使用“-3p”与“-5p”作为区分这两条序列的后缀替代旧的的命名法����。具体参考17.0版本出来时����,miRbase数据库的blog文章���:http://www.mirbase.org/blog/2011/04/mirbase-17-released/
2.如何筛选差异基因���?
可以在筛选的时候可能需要参考该参数���,一般如果专注于发现����,那么全部保留���,如果专注于差异表达����,可以仅保留high和middle拷贝的����。log2(Treat/Control)>0表示上调����,<0表示下调���,>1表示上调2倍����,<-1表示下调2倍�����。最简单的比较大小���,就是Treat-Control(看大于还是小于0)��,或者Treat/Control(看大于还是小于1)����,更有可读性的就是log2(Treat/Control)(>0表示上调���,<0表示下调��,>1表示上调2倍����,<-1表示下调2倍)负相关���、差异显著性pvalue值�����、foldchage���、最低拷贝值���。
全转录组测序
对组织或细胞在某一时刻或处理条件下转录出来的所有RNA进行测序分析���,全转录组的研究中包含对mRNA���、lncRNA���、circRNA���、miRNA进行分析�����,并结合多种RNA信息探索彼此之间潜在的调控网络����。
样本起始量与送样建议
|
样本类型 |
起始量 |
|
动物及临床脏器组织/脑组织等 |
>20mg |
|
动物及临床皮肤/骨/血管/脂肪组织等 |
>100mg |
|
植物叶片组织/花 |
>200mg |
|
植物根/茎/果实/种子 |
>500mg |
|
原代细胞/细胞系 |
>5×106个 |
|
中性粒细胞/嗜酸性粒细胞/嗜碱性粒细胞 |
>5×107个 |
|
外泌体样本 |
>1×108个 |
|
血清/血浆/脑脊液/关节积液/卵泡液 |
>2mL |
|
细胞培养上清液 |
>20mL |
|
尿液 |
>30mL |
|
总RNA |
>1μg且RIN>7.0 |
注意事项����:
① 组织样本建议保存在RNAlater�����、RNAHold����、RNAProtect等相关组织保存液中����,然后-80℃保存或干冰寄送���;
② 细胞样本使用TRIzol等裂解液充分裂解之后���,-80℃保存或干冰寄送���;
③ 更加详细的样本准备指南����,请联系在线客服����。
应用场景
应用场景1�����:结构分子招募核心蛋白与核酸
适用范围���:基础医学�����、生物化学与分子生物学等任意研究方向
lncRNA和circRNA有时候会作为一种特殊的RNA分子���,形成蛋白-蛋白复合物或蛋白-核酸的骨架分子���,对下游分子行使一系列调控作用����。如部分circRNA会招募一些泛素化酶对下游的蛋白行使降解功能����,或者部分lncRNA会招募一些转录因子与目的基因的启动子区域结合激活下游的基因表达等���。
应用场景2����:外泌体
适用范围���:临床与转化医学�����、基础医学等任意研究方向
lncRNA在外泌体中由于以碎片形式存在����,这种随机片段不仅不利于检测���,而且较低的浓度和较差的重复性����,使得lncRNA并不是外泌体研究的热门分子之一���。但是circRNA由于其稳定性��,除了从体液样本外泌体中分离circRNA可作为疾病标志物外�����,还能用于后期的外泌体治疗等领域����,是后ceRNA时代中较为热门的circRNA细分研究领域方向之一����。其中一个方向是疾病标志物���,一般以大样本队列研究居多����。一种是后期转化及机制研究方向����,包括外泌体受体细胞分子机制研究以及疾病治疗等方向����。
应用场景3����:翻译多肽
适用范围����:临床与转化医学��、基础医学����、生物化学与分子生物学等任意研究方向
circRNA上有一段未翻译的RNA序列���,称为IRES���,它可以折叠成类似于起始tRNA的结构����,并招募更多的核糖体���。正常情况下��,IRES不与eIF结合����,而是与一类称为ITAF的蛋白质结合���。ITAF的作用是将核糖体募集到circRNA的内部结构����,以启动蛋白质翻译���。此外��,还可能存在IRES增强子���,类似于circ-ZNF609的UTR元件��。也有研究通过翻译组测序与转录组测序����,发现了非编码RNA LINC-PINT形成环状RNA后可被翻译形成功能多肽����。
应用场景4��:ceRNA调控机制
ceRNA全称competing endogenous RNA���,是一种能够竞争结合RNA的作用元件���。通常lncRNA和circRNA会竞争结合miRNA����,我们一般把lncRNA和circRNA可以称作ceRNA����。ceRNA调控网络全称ceRNA regulation network����,指的是有ceRNA参与的整个调控网络cascade���。而ceRNA分析指的是对整个ceRNA调控网络进行分析���。一般有circRNA-miRNA-mRNA分析或lncRNA-miRNA-mRNA分析���。
案例展示
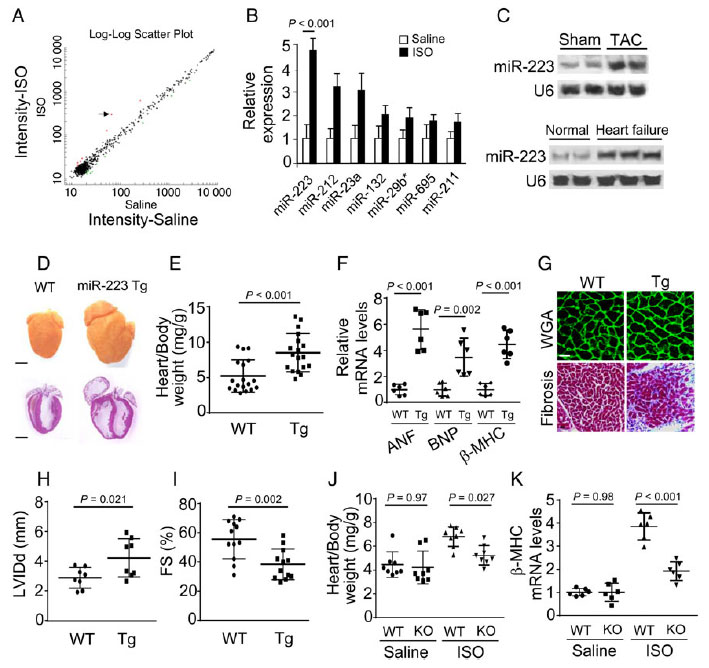
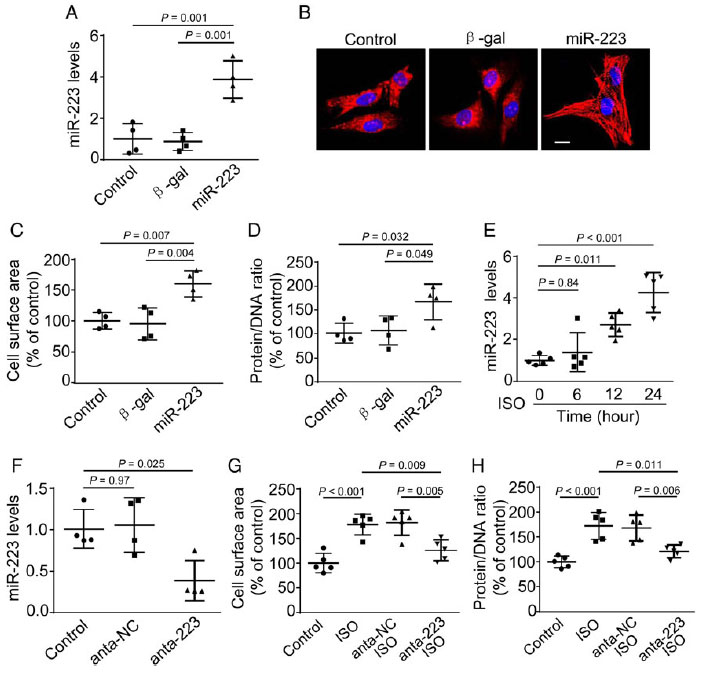
circular RNA(HRCR)靶标结合miR-223保护心脏免受心肌肥厚和心脏衰竭
本研究中作者首先通过miRNA基因芯片分析����、Northern blot���、RT-PCR等技术手段发现��,ISO处理14天后��,miRNA-223的表达量显著上调��,并且在患有心脏肥厚和心脏衰竭的人和小鼠心脏中miRNA-223的表达量也有明显的上调���,初步得出结论���:miRNA-223参与调控心肌肥厚和心脏衰竭��。接下来作者利用miR-223过表达转基因小鼠以及miR-223基因敲除转基因小鼠进一步验证表型��,结果发现miR-223过表达小鼠成年后出现明显的心肌肥厚和心脏衰竭症状���,而miR-223基因敲除小鼠表型相反����,进一步证明在活体内���,miR-223可以诱导心肌肥厚和心脏衰竭�����。
接下来作者进一步研究miR-223对心肌细胞的影响�����,结果发现miR-223过表达细胞中出现明显的心肌肥大反应����,而miR-223敲除细胞中����,ISO处理引起的心肌肥大反应明显被破坏���,说明miR-223过表达会促进心肌肥厚��,而miR-223基因敲除会阻断心肌肥厚���。
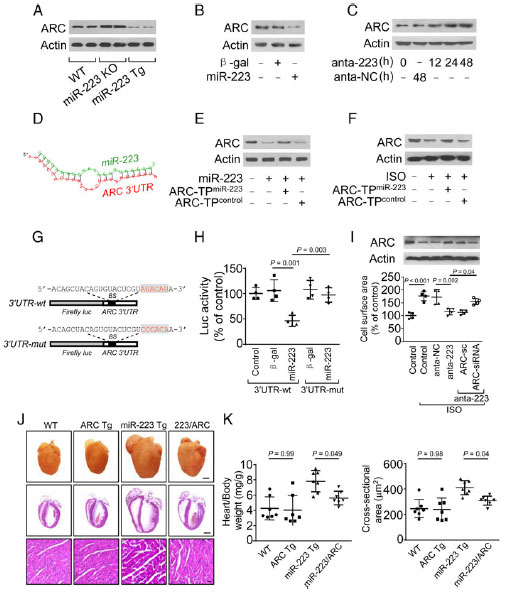
为了进一步研究miR-223调控心肌肥厚和心脏衰竭的分子机制��,作者筛选了一些参与调控心肌肥厚的基因作为miR-223的靶标候选基因����,然后通过western blot分析miR-223过表达和缺失突变体中上调以及下调的基因��,结果发现miR-223可以显著抑制ARC的表达���,而前人已经有研究报道ARC参与调控心肌肥厚和细胞凋亡���,所以初步推断ARC可能是miR-223下游的靶标位点����。
CircRNA和miRNA的互作是目前研究的一个热点话题���,为了进一步研究是否有CircRNA参与调控miR-223的表达�����,作者从CircRNA数据库中随机筛选出100个CircRNA�����,通过qRT-PCR验证筛选到36个CircRNA���,然后通过Northern blot筛选到一个特异性表达的CircRNA: mm9-circ-012559(HRCR)����。
然后作者通过RNAhybrid分析发现HRCR包含6个CircRNA的靶标结合位点����,初步表明HRCR可以和miR-223结合����。接下来作者通过生物素标记���,qRT-PCR����,Northern bolt�����,pull down assay���、荧光原位杂交(FISH)等技术手段��,从正向以及反向两个角度分别验证HRCR和miR-223之间的互作关系��,结果发现miR-223和HRCR在细胞质中存在共定位现象����,HRCR可以直接结合到miR-223上����。
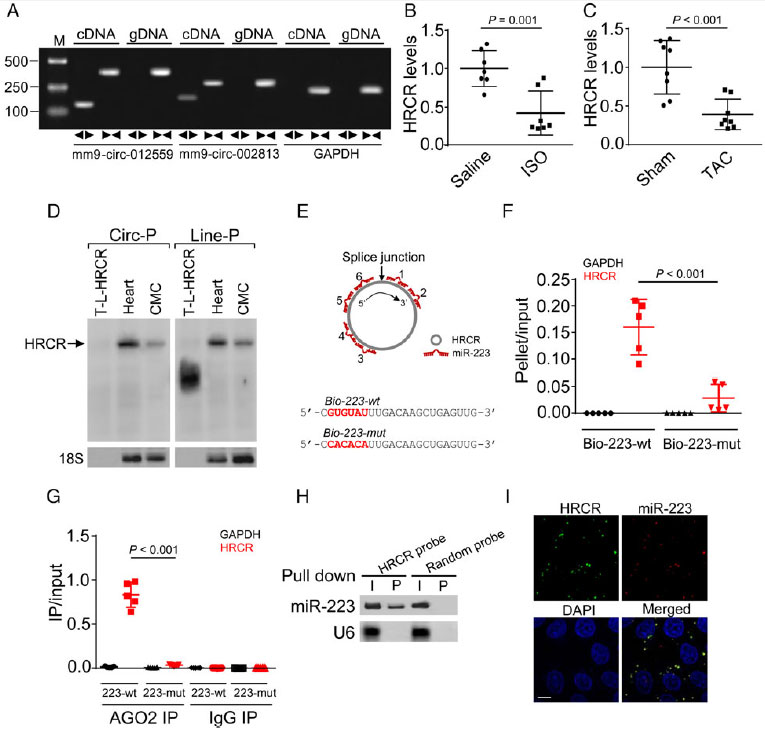
HRCR和miR-223存在互作���,而miR-223可以通过调控下游ARC的表达��,进而调控心肌肥厚和心脏衰竭��,为了进一步研究HRCR对ARC表达的影响���,作者构建了HRCR过表达载体和基因敲除突变体���,结果发现HRCR过表达载体中�����,ARC的表达量和活性显著增加��,HRCR敲除突变体中���,ARC的表达量和活性显著降低��,并且HRCR可以消除miR-223对ARC表达的抑制作用��,说明HRCR可以通过调控miR-223以及ARC的表达��,进而调控心肌肥厚���。
参考文献
Wang K, Long B, Liu F, et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223.[J]. European Heart Journal, 2016, 37(33):2602-2611.
常见问题
1.全转录组测序为什么需要构建两个文库
全转录组测序需要构建2个测序文库����,一个小RNA文库和一个去除核糖体RNA的链特异性文库��,然后分别上机测序���。小RNA长度较短��,一般小于50nt��,采用测序策略为50SE��。其他三类RNA序列一般大于200��,通常在1000以上����,最高可达几万��,通过片段化构建150PE测序文库���。最后小RNA文库可以获得miRNA序列信息�����,去核糖体的链特异性文库可以获得mRNA����、lncRNA和circRNA的序列信息���。
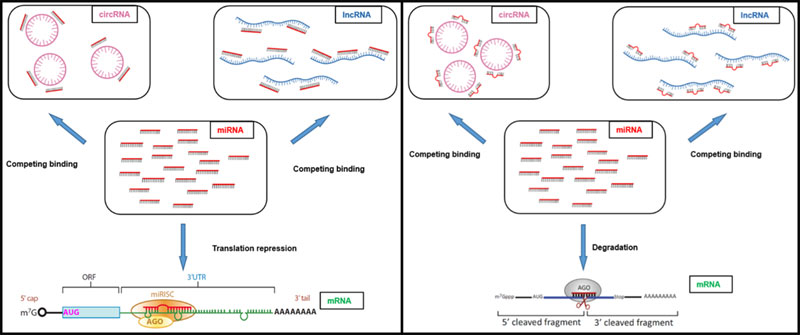
2.什么是ceRNA调控网络(ceRNA networks)��。
这些非编码RNA如lncRNA和circRNA等会竞争结合miRNA��,导致miRNA调控的靶基因发生变化���,最终体现在蛋白的表达水平上��,而miRNA处于ceRNA调控网络中的核心地位��。如下图
原核转录组
基于高通量测序平台���,构建链特异性文库��,研究原核生物在某个时期或者在某种环境条件下转录出来的所有mRNA����。
样本起始量与送样建议
|
样本类型 |
起始量 |
|
细菌 |
>200 mg |
|
总RNA |
>1μg且RIN>7.0 |
注意事项���:
① 建议收集对数生长期的样品送样���;
② 将样本放入液氮预冷的无酶管����,ddH2O或灭菌的PBS后���,-80℃保存或干冰寄送����;
③ 更加详细的样本准备指南����,请联系在线客服��。
翻译组
经典中心法则指出DNA转录产生mRNA���,而mRNA指导蛋白质的合成���,蛋白质是生命活动的主要承担者�����。然而科学家们发现人类基因组中70%以上的DNA可以转录产生RNA而具有编码蛋白能力的mRNA只占3%左右��,说明多数转录的RNA分子都属于非编码RNA����,对于RNA的翻译主要分为��:不翻译����,部门翻译��,从头翻译��,过度翻译��。
随着NGS的广泛应用��,翻译组学研究迅速成为现代生命科学的热点之一���,其中��,核糖体印迹测序技术是该研究的利器之一(Ribo-seq)�����,该技术由Weissman课题组于2009年发表在Science杂志上����。此后相继应用到微生物�����、酵母�����、小鼠�����、人类组织����、玉米���、拟南芥����、斑马鱼等多个物种研究上���。